很多人搜 scRNA-seq 分析流程,并不是想看一张漂亮流程图,而是想知道真实项目到底怎么推进。项目刚启动时,最怕的不是步骤多,而是每一步之间接不上。前面做了不少预处理,后面却不知道该怎么接到分析目标上,这种情况很常见。
这篇文章按项目推进顺序,把常见的 scRNA-seq workflow 拆开讲。每一步我都会尽量回答 3 个问题:它在解决什么、输入输出是什么、哪里最容易出错。
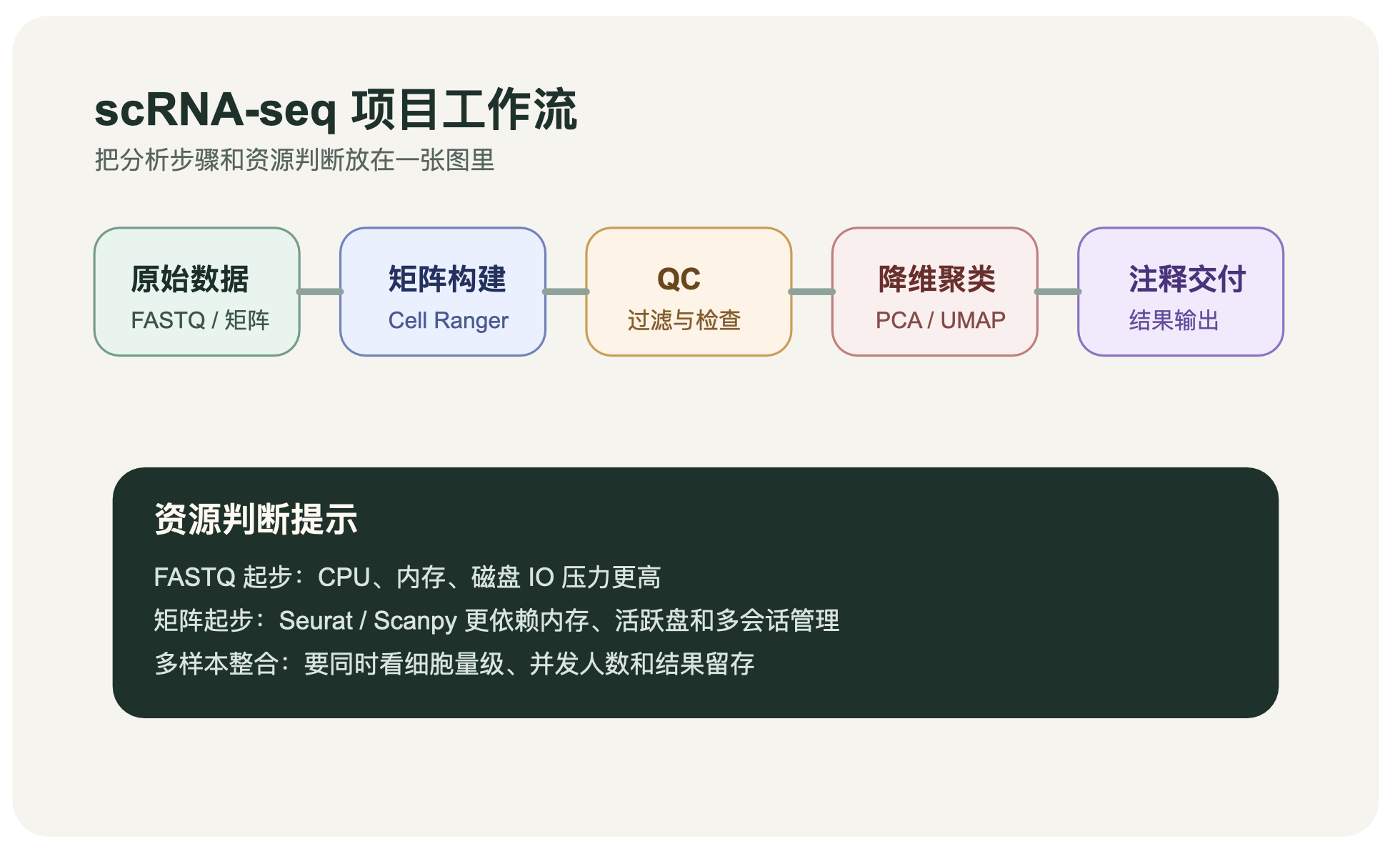
scRNA-seq 分析流程先看什么
正式开始前,先确认 3 件事:
你的数据从
FASTQ、表达矩阵还是对象文件开始这次分析是单样本探索,还是多样本比较
最终目标是出图、写文章,还是做可复现交付
这 3 件事会直接影响流程长度、资源压力和后面的解释方式。
第一步:明确数据来源和实验策略
你手上的数据一般来自下面几类:
自己送测的单细胞项目
GEO、SRA 等公共数据库
合作方直接给的矩阵或对象文件
起点不同,scRNA-seq 分析流程就不会完全一样。
如果拿到的是 10x Genomics 原始数据,常见做法是先通过 Cell Ranger 生成表达矩阵;如果拿到的是已经整理好的 matrix,就可以直接进入 Seurat 或 Scanpy 阶段。
这一段别嫌啰嗦。很多项目后面反复返工,问题就出在最开始没说清数据起点。
![生信分析跑不动?试试稳定高性价比的生信云 → [www.tebteb.cc]](https://www.tebteb.cc/upload/%E7%94%9F%E4%BF%A1%E5%9C%86%E6%A1%8C-kgzw.png)
第二步:原始数据处理与矩阵构建
这一段主要发生在表达矩阵生成之前,目标是把原始 reads 变成“基因 x 细胞”的计数矩阵。
常见步骤包括:
reads 质控
barcode 识别
UMI 去重
比对到参考基因组或转录组
生成 feature-barcode matrix
对 10x 数据来说,Cell Ranger count 是最常见的起点。它的输出通常包括:
matrix.mtx.gzfeatures.tsv.gzbarcodes.tsv.gz
后续单细胞分析基本就是从这几类文件接下去。
第三步:细胞与基因质控
QC 是整个 scRNA-seq 分析流程里最不能草率的一步。cluster 不稳定、线粒体比例异常、双细胞太多,这些问题往往都能往前追到质控阶段。
单细胞质控时,最常看的指标是:
每个细胞检测到的基因数
nFeature_RNA每个细胞总计数
nCount_RNA线粒体基因比例
percent.mt
一般不要上来就套固定阈值。更稳的做法是先看分布,再判断过滤线。比如:
检测到的基因数太低,可能是低质量细胞或空液滴
UMI 数过高,可能是双细胞
线粒体比例过高,往往提示细胞状态不好
不同组织、不同平台、不同样本质量下,阈值差异会很大。照搬别人的 QC 线,是新手特别容易踩的坑。
第四步:标准化与高变基因筛选
过滤掉低质量细胞之后,通常会做标准化。原因很简单,不同细胞测到的总 counts 不一样,不能直接横着比。
常见处理包括:
library size normalization
log transformation
筛选高变基因
高变基因这一步很关键。不是每个基因都能帮助你区分细胞群,真正携带结构信息的往往只是其中一部分。把这些基因挑出来,后面的 PCA 和聚类才更稳。
第五步:降维
scRNA-seq 数据维度很高,直接在原始表达矩阵上做结构识别既慢也不稳,所以通常会先降维。
常见顺序是:
先做 PCA
再基于 PCA 结果构图和聚类
最后用 UMAP 或 t-SNE 做二维可视化
PCA 更多是在保留主要变异信息,UMAP 更多是为了展示结构。UMAP 图很好用,但它不是统计结论本身,这一点最好早点建立。
第六步:邻近图构建与聚类
降维之后,通常会基于主成分结果构建 KNN 图,再进一步做聚类。目标是把表达模式相近的细胞分成不同群体。
聚类时最常被提到的参数是 resolution。它会影响分群粗细:
分辨率太低,不同群体可能被并在一起
分辨率太高,同一类细胞可能被切得过碎
所以聚类不是“跑出结果就完了”,而是要结合 marker、样本来源和研究问题一起判断是否合理。
第七步:细胞类型注释
聚类完成后,最核心的一步就是 annotation,也就是给 cluster 贴标签。
常见做法包括:
根据经典 marker gene 手动注释
参考公开数据库辅助判断
用自动注释工具先做初筛,再人工复核
举个常见例子:
CD3D、IL7R高表达,通常提示 T 细胞MS4A1高表达,常见于 B 细胞LYZ、S100A8较高,往往提示髓系细胞
自动注释能提速,但最终判断还是要回到文献、样本来源和课题背景。
第八步:差异表达分析
当 cluster 或细胞类型确定之后,很多项目就会进入比较阶段。常见问题包括:
某个 cluster 相比其他 cluster 的 marker gene 是什么
疾病组和对照组中,同类细胞之间有什么差异
处理前后特定细胞群是否发生表达重塑
这一步最需要小心的,是不要把所有细胞不加区分地混在一起比较。真正严谨的分析,通常会同时考虑细胞层面和样本层面的解释。
第九步:富集分析与下游功能分析
差异基因出来之后,常见的延伸分析包括:
GO / KEGG 富集分析
GSEA
轨迹分析
细胞通讯分析
拟时序分析
转录因子活性分析
不是每个项目都要把这些做满。围绕核心问题往下挖,通常比“每个模块都来一点”更有价值。
第十步:结果可视化与交付
一套完整的 scRNA-seq 分析流程,最后要能交付别人看得懂、自己以后也能复用的结果。
常见交付内容包括:
QC 图
UMAP / t-SNE 图
marker gene feature plot
heatmap
violin plot
差异基因表
注释结果表
分析脚本或 notebook
如果是服务型项目,最好再补上:
参数记录
软件版本
样本信息表
过滤标准说明
这会直接影响后续复盘、返修和论文写作效率。
不同起点对服务器压力有什么区别
“流程图看懂了”和“机器跑得动了”是两回事。尤其是下面这几种起点,资源压力差异很明显:
从
FASTQ开始:通常要跑Cell Ranger或类似流程,CPU、内存和磁盘 IO 压力都比较大从表达矩阵开始:前处理少一些,但 Seurat 或 Scanpy 阶段会更吃内存
从多样本整合开始:如果还涉及批次校正和多人并发,内存和高速存储通常更先成为瓶颈
如果你现在已经在搭实验室环境、做生信服务或准备采购机器,建议别只看步骤名,要把每一步对应的数据规模和并发需求一起估算清楚。
scRNA-seq 分析流程常见问题
scRNA-seq 分析一定要从 FASTQ 开始吗
不一定。很多公共数据可以直接从表达矩阵开始,合作方也可能直接给 Seurat object。关键是先确认手上的数据起点。
scRNA-seq 质控阈值有固定标准吗
没有。QC 阈值通常要结合组织类型、测序深度和样本质量判断,最好先看分布再定线。
scRNA-seq 聚类后就算结束了吗
远远不算。聚类只是结构识别的开始,后面还要做注释、差异分析和研究问题解释。
结语
一条靠谱的 scRNA-seq 分析流程,不是把所有工具都堆上去,而是知道每一步为什么存在、输入输出是什么、哪些结果可以解释、哪些地方还需要回头核验。主线一旦吃透,后面不管你是换 Seurat、换 Scanpy,还是做更复杂的整合分析,都会有底。