在中文单细胞分析语境里,Seurat 基本绕不过去。它流行不是偶然,核心原因很简单:常见流程它都串起来了,资料多,案例也多,拿来入门和做项目都方便。
但问题也很典型。很多人学 Seurat,是把代码段复制过去跑一遍;一旦换数据、换过滤条件、换样本数量,就开始发懵。真正难的不是函数本身,而是你不知道什么时候该停下来判断。
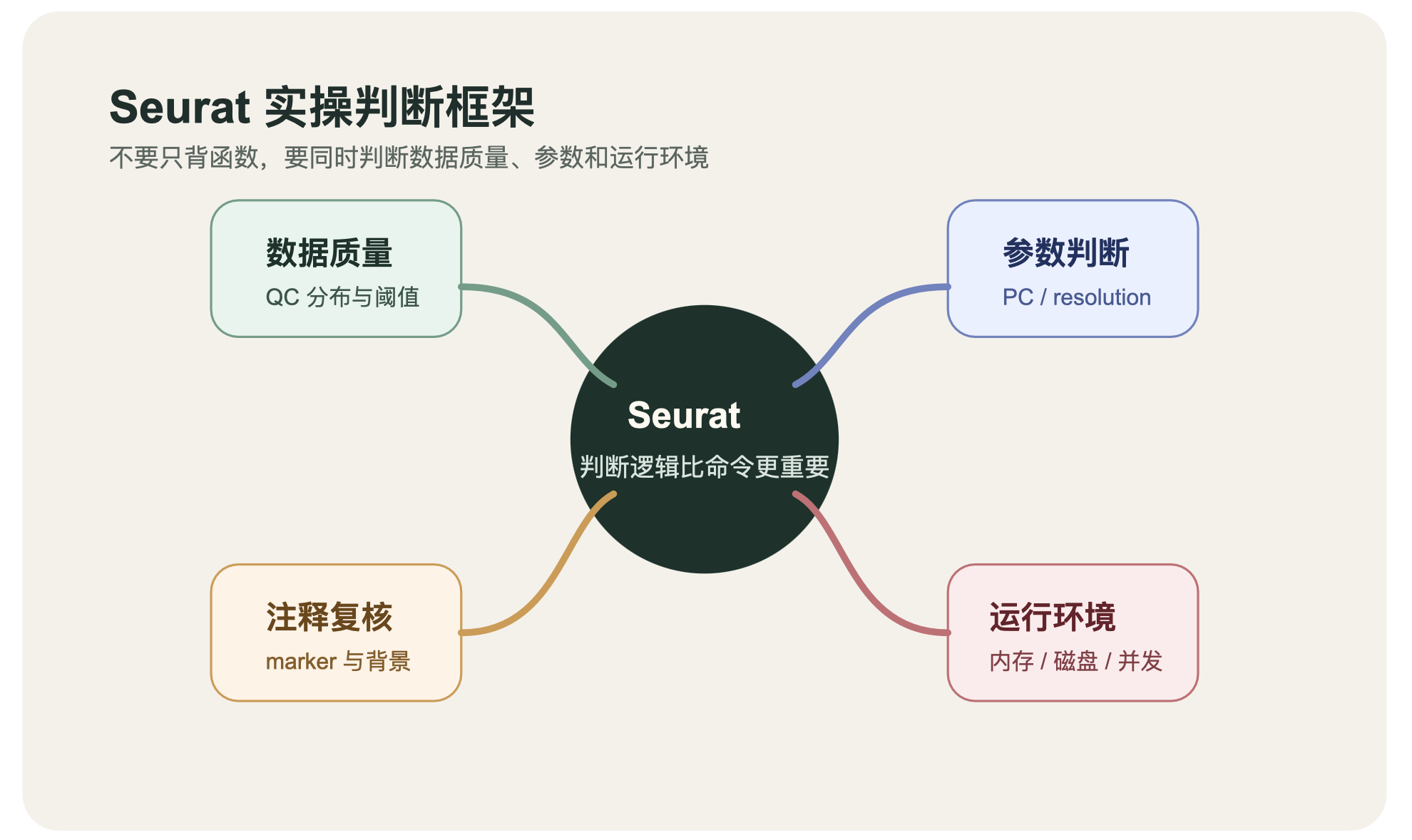
这篇 Seurat 教程不想只给你命令列表,更想把实操时真正有用的判断逻辑讲清楚。
Seurat 是什么,适合做什么
Seurat 是一个基于 R 的单细胞分析框架,适合处理下面这些常见任务:
读取单细胞表达矩阵
做细胞和基因质控
标准化和高变基因筛选
PCA、UMAP、t-SNE 降维
聚类分析
marker gene 查找
细胞类型注释
多样本整合分析
如果你是新手,Seurat 的优势很现实:教程多,社区成熟,遇到问题更容易搜到可参考的思路。
![生信分析跑不动?试试稳定高性价比的生信云 → [www.tebteb.cc]](https://www.tebteb.cc/upload/%E7%94%9F%E4%BF%A1%E5%9C%86%E6%A1%8C-kgzw.png)
一个典型的 Seurat 分析流程
Seurat 教程里最常见的主线通常是这样:
创建对象
计算 QC 指标并过滤
数据标准化
找高变基因
数据缩放
PCA
FindNeighbors()+FindClusters()RunUMAP()FindAllMarkers()注释细胞类型
这条主线和标准 scRNA-seq workflow 基本一致。Seurat 的价值主要在于把这些步骤组织得很顺,适合你先建立节奏感。
Seurat 最关键的不是函数名,是判断逻辑
1. CreateSeuratObject 只是起点
很多教程在 CreateSeuratObject() 之后立刻往下跑,但真实项目里,你最好先确认三件事:
矩阵是否完整
基因名是否规范
样本信息是否已经准备好
前期元数据没整理好,后面做分组比较、批次检查和下游分析时会非常别扭。
2. QC 阶段先看分布,不要先背阈值
Seurat 里常做的质控指标包括:
nFeature_RNAnCount_RNApercent.mt
这些数值没有统一答案。比如线粒体比例,在肿瘤样本、外周血和冷冻组织里,经验阈值就可能完全不同。
所以更成熟的做法是先看分布,再决定过滤线,而不是上来就照抄“线粒体比例大于 10% 全删掉”。
标准化和高变基因为什么重要
标准化的目的是减少不同细胞测序深度差异带来的影响。高变基因筛选的目的是把真正有助于区分细胞状态和细胞类型的信息提取出来。
如果这一步做得糊里糊涂,后面的 PCA 和聚类结果通常也会飘。
对初学者来说,先记住一句话就够了:不是每个基因都值得带进后续结构分析,真正重要的是那些在细胞之间差异更明显的基因。
PCA、聚类和 UMAP 在 Seurat 里分别解决什么问题
这是很多新手最容易混的一段。
PCA
PCA 用来压缩维度,提炼主要变化方向。它更像是后续分析的基础表示层。
聚类
聚类是在低维空间里把表达模式相似的细胞归成不同群体。Seurat 里通常通过邻近图和社区发现算法完成。
UMAP
UMAP 是把高维结构映射到二维平面,方便可视化。它很适合看大致分群,但不要把 UMAP 上的视觉距离直接当成真实生物学距离。
FindAllMarkers 之后,怎么注释更稳
FindAllMarkers() 是 Seurat 教程里出镜率很高的函数,因为它能帮你识别每个 cluster 的代表基因。但 marker 列表出来后,真正的判断才刚开始。
更稳妥的注释思路通常是:
先看 cluster 的 top markers
再看多个经典 marker 是否共同支持同一结论
结合样本来源、组织类型和研究背景做复核
比如在免疫微环境数据里:
CD3D、TRBC1、IL7R一起升高,更像 T 细胞MS4A1、CD79A、CD74上升,更像 B 细胞
真正的注释,最好别只靠一个基因拍板。
Seurat 项目里最常见的几个问题
问题一:聚类太碎或太粗
这通常和下面几个因素有关:
QC 阈值不合理
使用的 PC 数量不合适
resolution偏高或偏低样本批次效应没有处理
问题二:不同样本混在一起,看不出真实结构
这往往提示你要检查批次效应,或者考虑样本整合流程,而不是把所有细胞简单拼起来直接聚类。
问题三:注释很难下结论
这不一定是你不会看 marker,也可能是:
cluster 内部本身异质性强
分辨率不合适
数据质量一般
样本里存在过渡状态细胞
换句话说,注释困难有时是生物学现实,不一定是分析失败。
Seurat 跑得慢,很多时候不是代码问题
不少用户把 Seurat 卡顿、报错、运行慢,直接理解成“代码写错了”。但真实项目里,下面这些原因更常见:
单个对象过大,本地电脑或低内存环境扛不住
多样本整合时多个对象同时驻留内存
活跃项目和原始数据都堆在慢盘上,IO 成为瓶颈
团队共用一台机器时,多个会话同时运行,资源抢占明显
这也是为什么很多人能在教程数据上顺利跑通 Seurat,换到真实项目却频繁卡住。学习成本是一回事,运行环境能不能跟上是另一回事。
Seurat 常见问题
Seurat 适合单细胞分析新手吗
适合。它的资料丰富、案例多,适合拿来建立完整分析主线。
Seurat 能直接当黑箱用吗
不建议。你可以先靠它入门,但越往后做,越需要理解每一步输入输出和参数调整逻辑。
Seurat 对服务器配置要求高吗
中小型数据在本地也能跑,但一旦进入多样本整合、较大细胞量级或团队并发使用,内存和存储很快就会成为限制。可以配合这篇看:单细胞分析服务器配置怎么选:CPU、内存、存储与部署建议。