单细胞分析这几年确实很热,但对新手来说,第一感受通常不是“先进”,而是“信息太碎”。教程里一会儿是 10x Genomics,一会儿是 UMI、barcode、QC、PCA、UMAP、cluster、annotation,再加上 Seurat 和各种图,刚看没多久就容易乱。
如果只记一句话,我会这样说:单细胞分析就是把原来混在一起看的平均信号,拆到单个细胞层面,再看不同细胞群之间到底有什么差异。
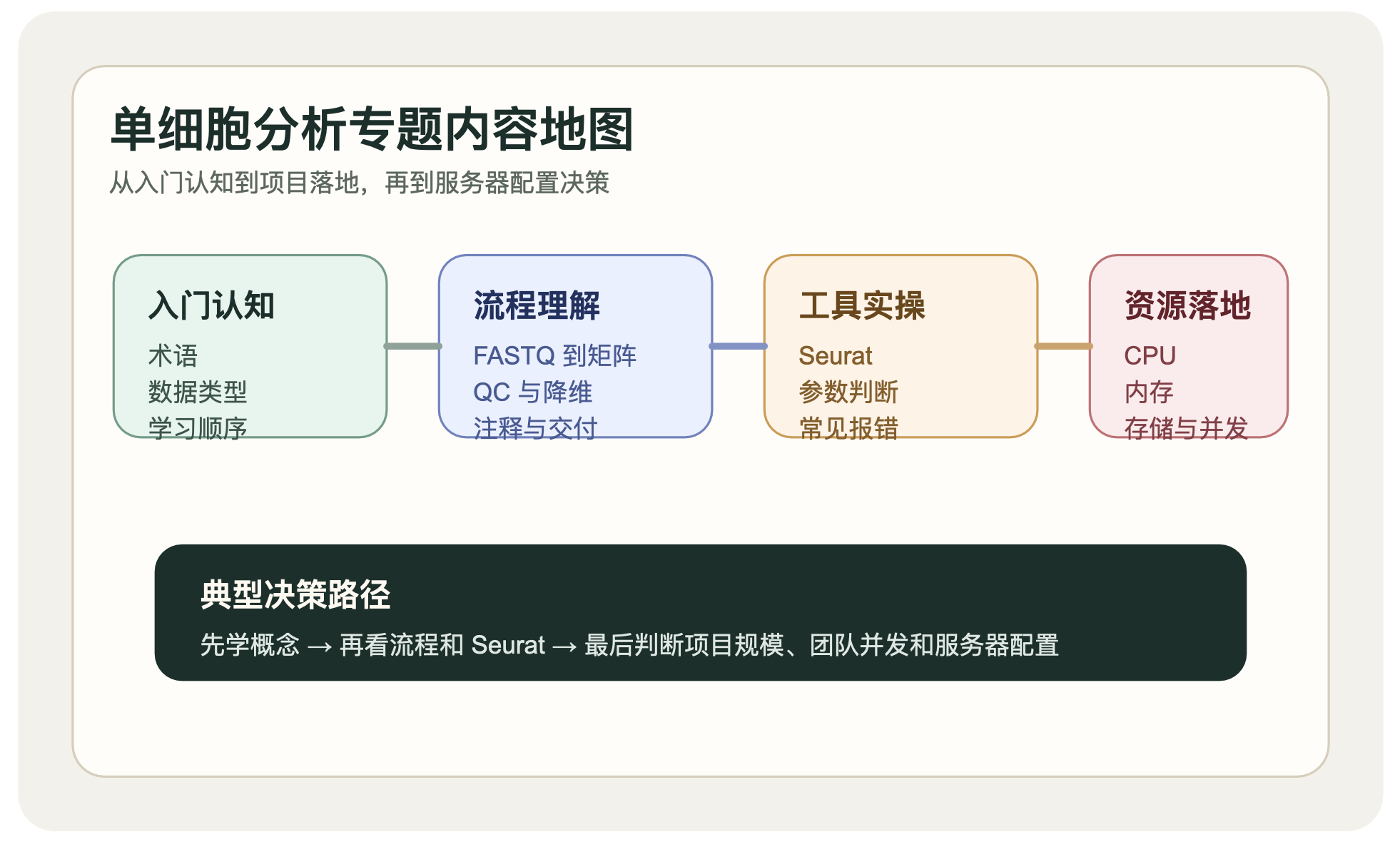
这篇文章不打算一上来讲复杂算法,而是先帮你把框架搭起来。框架一旦顺了,后面的流程、代码和图就没那么吓人了。
单细胞分析是什么
大多数人说“单细胞分析”,实际最常指的是 scRNA-seq,也就是单细胞转录组测序分析。它的目标很直接:在单细胞分辨率下观察基因表达差异。
和传统 bulk RNA-seq 相比,最大的区别是你不再只看到一个样本的平均表达,而是能看到样本内部不同细胞群的异质性。以前被平均值抹平的东西,现在能重新冒出来。
这类分析常用来回答下面这些问题:
一个组织里到底有哪些细胞类型
是否存在稀有细胞群
某类细胞在疾病状态下发生了什么变化
细胞是否处在分化、激活或应激过程中
细胞之间可能有哪些通讯关系
所以,单细胞分析的价值不只是“更细”,而是它能把以前看不见的结构重新拉出来。
单细胞分析最常见的数据类型
新手先别急着把所有单细胞组学都装进脑子里。先记住下面三类就够用了:
scRNA-seq:看单细胞基因表达,是最常见的入门方向snRNA-seq:看单核转录组,常用于不容易获得完整细胞的组织spatial transcriptomics:既看表达,也保留空间位置信息
如果你现在的目标是“先入门单细胞分析”,重点先放在 scRNA-seq 就行。大多数教程、案例和讨论,默认也都是围绕它展开。
单细胞分析标准流程是什么
很多人觉得单细胞分析难,是因为看到的总是零散步骤。换成流程视角,其实会清楚很多。
一套常见的 scRNA-seq 流程通常包括:
原始数据获取
表达矩阵构建
细胞和基因质控
标准化与高变基因筛选
降维
聚类
细胞类型注释
差异表达分析
富集分析或其他下游分析
你可以把它看成一条从原始 reads 走到生物学解释的路径。前半段更像是整理数据、控制噪声,后半段才逐渐进入结构识别和结果解释。
如果你想看展开版,可以继续读这篇:scRNA-seq 分析流程详解:从原始数据到细胞注释。
新手最容易混淆的几个术语
Barcode
barcode 是区分不同细胞来源的标签。因为很多细胞会混在一起上机,后面要靠 barcode 把 reads 分回各自的细胞。
UMI
UMI 是分子标签,主要用来减少 PCR 扩增带来的偏差。简单理解,它帮助我们更接近真实转录本数量。
Feature-barcode matrix
这是单细胞分析真正开跑的地方。矩阵的行通常是基因,列是细胞,数值是某个细胞里某个基因的表达计数。
QC
QC 就是质控,主要用来筛掉低质量细胞和明显噪声。最常看的指标包括:
每个细胞检测到的基因数
每个细胞总 UMI 数
线粒体基因比例
PCA / UMAP
单细胞数据维度很高,不能直接拿原始矩阵去看结构,所以要先降维。PCA 更多是为后续分析服务,UMAP 更多是为了可视化展示。
Cluster
cluster 就是聚类结果,代表一群表达模式相近的细胞。后续注释通常也是先看 cluster,再判断它可能对应什么细胞类型。
Annotation
annotation 是细胞类型注释,也就是根据 marker gene、参考数据库和已有文献知识,给每群细胞贴标签。
单细胞分析入门该怎么学
新手最容易走偏的一点,是过早开始背命令。更顺的学习顺序通常是下面这样。
1. 先搞清分析目标
你要知道自己为什么做单细胞分析。是为了找细胞亚群、看状态变化、研究肿瘤微环境,还是想做细胞通讯?目标不同,后面关注的重点也会变。
2. 再理解标准流程
先别急着背函数,先理解每一步在解决什么问题。比如为什么要做质控,为什么要筛高变基因,为什么聚类前要先降维。
3. 再学一个主工具
对中文用户来说,Seurat 依然是比较合适的入门工具。资料多、案例多、社区成熟,用它先建立完整分析思路,成本相对低一些。
你可以接着看这篇:Seurat 教程:单细胞数据分析的标准实操思路。
4. 最后再补高级分析
像批次效应校正、轨迹分析、细胞通讯、SCTransform、多样本整合这些内容,放在基础流程跑通之后再学更合适。太早进去,通常只会让你更乱。
单细胞分析入门常见误区
误区一:代码能跑就是会了
这是最常见的一类。代码能跑出来不代表你理解了结果。比如 cluster 为什么这么分、线粒体比例为什么要过滤、marker gene 为什么能支持注释,这些要是说不清,说明只是流程跑通了,还没真正入门。
误区二:默认参数就是标准答案
默认参数只是起点。不同组织、不同测序深度、不同样本质量下,QC 阈值、PC 数量和聚类分辨率都可能要调。
误区三:一开始就拿超大数据集练手
几十万细胞的数据当然更“真实”,但不适合入门。更稳的做法是先找一个 3k 到 20k 细胞量级的数据,把完整流程走通。
误区四:图画出来就算分析结束
UMAP 只是入口,不是结论。真正有价值的是:这些细胞群是谁、为什么会分开、差异背后可能说明什么。
学单细胞分析需要什么基础
不用把门槛想得太高,但有几块基础最好心里有数:
生物学基础:知道基因表达、细胞类型和 marker 的基本概念
R 基础:能读脚本、装包、处理数据框
Linux 基础:至少能在服务器上管理文件、跑基础命令
统计基础:知道差异分析和多重检验在说什么
这不是说你必须一开始全会,而是你要知道后面要补哪些地方。
从教程到真实项目,最容易卡在哪
很多人看完入门内容之后,会以为下一步只是继续学更多 Seurat 函数。其实真正从“看懂教程”走到“能跑项目”,最常卡住的往往是这些地方:
你拿到的数据起点并不统一,可能是
FASTQ、矩阵,也可能是对象文件练手数据和真实项目的数据规模完全不是一个量级
课题或服务项目往往要求保留脚本、参数和中间结果,不是只出几张图
本地电脑能跑通,不等于团队共享服务器也能稳定运行
这一段如果你不提前补,等项目真开始,问题一般会一起冒出来。
快速回答:单细胞分析新手先学什么
先学概念和流程,再学 Seurat。
先理解 QC、降维、聚类和注释各自解决什么问题,再背函数。
先用中小型数据集把一遍流程走通,再碰大项目和复杂下游分析。